
Understanding the Ngs File Format: A Comprehensive Guide
In the world of genomics, the Ngs file format plays a crucial role in storing and analyzing Next-Generation Sequencing (NGS) data. Understanding this file format is essential for researchers and scientists working with large genomic datasets. In this comprehensive guide, we will explore the ins and outs of the Ngs file format , different types of NGS file formats, and best practices for managing NGS data effectively.
Introduction to the NGS File Format
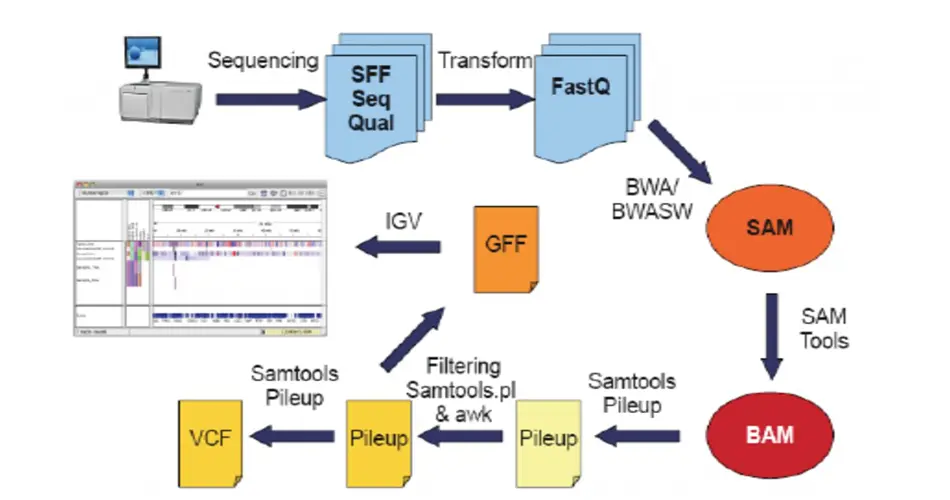
The NGS record arrangement is essential for putting away the storm of information delivered by Next-Generation Sequencing innovations. This organization typifies the substance of DNA or RNA sequences determined from cutting-edge sequencing strategies. Its design is meticulously optimized for both the compact capacity and practical processing of voluminous genomic information. As the spine of genomic information examination, the Ngs record arrangement underpins the effective dealing with and elucidation of sequencing data, encouraging headways in genomics research and personalized medication. Understanding its structure and utility is foundational for researchers and analysts exploring the complexities of genomic datasets.
Types of NGS File Formats
Within the realm of genomic research, various NGS file formats are utilized to accommodate the diverse stages of data analysis. Among the most widely recognized are FASTQ, BAM, and VCF files, each serving distinct roles in the sequencing workflow. FASTQ files are essential for capturing raw sequencing reads alongside their quality scores, providing the foundation for initial data assessment. BAM files, on the other hand, are integral for storing alignment information, offering insights into how sequences compare to a reference genome. Lastly, VCF files play a pivotal role in variant calling, documenting genetic variations identified during analysis. These formats collectively streamline the process from raw data collection to the elucidation of genetic variances, underpinning the intricate pipeline of NGS data examination. Understanding the specific function and application of each file format is indispensable for researchers aiming to navigate the complexities of genomic sequencing data effectively.
Working with FASTQ Files
To handle FASTQ files effectively, one must engage in quality assessment and enhancement processes right at the outset. These files encapsulate both the nucleotide sequences and their corresponding quality scores - a crucial aspect for ensuring the integrity of data before proceeding to more complex analysis stages. The primary steps involve scrutinizing the quality scores to identify and exclude low-quality reads, as well as trimming sequences to remove adapter sequences or other non-biological artifacts.
Tools such as FastQC and Trimmomatic are frequently employed for these purposes, allowing researchers to evaluate and refine their datasets by filtering out undesirable elements. This preparatory work lays a solid foundation for the accurate alignment and subsequent analysis of sequencing data, ensuring that only the highest quality reads are considered for further study. Engaging with FASTQ files through these initial quality control measures is a critical step in the NGS workflow, setting the stage for reliable and meaningful genomic analysis.
Aligning Sequences and Understanding BAM Files
Upon completing the initial quality control of FASTQ files, aligning the sequences against a reference genome represents a pivotal phase in NGS data processing. This critical step is captured within BAM files, which serve as a repository for the aligned sequences, detailing their respective positions on the genome and the quality of each alignment. The ability to interpret BAM files is essential for further analyses such as variant calling. These files provide a comprehensive view of how individual sequences align with the reference, facilitating the identification of discrepancies that could indicate genetic variations. Mastery of navigating and analyzing BAM files is a fundamental skill for researchers, enabling them to accurately assess the quality of alignments and to proceed confidently into deeper genomic investigations.
Variant Calling and the VCF File Format
Variant calling may be a basic stage in NGS information analysis where genetic differences from a reference sequence are identified. The Variant Call Arrange (VCF) serves as the vehicle for reporting these genetic varieties, including single nucleotide polymorphisms (SNPs) and insertions/deletions (lingers). A VCF records points of interest to the particular areas, characteristics, and certainty scores of recognized variants, making it a vital device for hereditary inquiry. It allows scientists to pinpoint and catalog genetic mutations, offering a window into the underlying genetic factors of diseases or traits. Navigating the complexities of VCF files is pivotal for extracting meaningful insights from sequencing data, enabling the pursuit of more targeted and informed genomic studies.
Best Practices for Managing NGS Data
Successful management of Ngs file format information is vital for ensuring inquiry about judgment and encouraging smooth investigations. Key strategies incorporate receiving an efficient approach to record organization by utilizing significant naming traditions that reflect the substance and arrangement of examination, which helps in quick identification and decreases confusion. It is additionally basic to preserve point by point documentation of the analytical processes, counting the program and parameters utilized, to upgrade reproducibility and streamline collaboration. Furthermore, actualizing a strong reinforcement strategy is basic, including both nearby and cloud-based arrangements, to protect against information loss and guarantee information life span. Frequently overhauling and curating your information, while keeping an organized record of record forms, can avoid excess and keep up a clean dataset for ongoing and future projects. Following to these hones not as it were optimizes the inquiry about workflow but also fortifies the unwavering quality and availability of genomic information.
Conclusion
Acing the Ngs file format record arranged is vital for those diving into the vast and complex world of genomics investigate. An exhaustive get handle on the different NGS record groups, such as FASTQ, BAM, and VCF, prepares analysts with the instruments vital to exploring the complexities of genomic sequencing information. Executing the best hones for information administration guarantees the judgment and viability of the research preparation. As the field of genomics proceeds to advance, remaining educated and versatile in modern techniques will be key to opening the complete potential of NGS information. Keep an eye out for assistance experiences and techniques that will help in tackling the control of genomics to drive logical disclosure.









